
About me
I am a Computational/Medicinal/Analytical chemist based in Augbsurg, Bayern. More about research.
In my career, I’ve delved into a diverse array of projects that have fueled my passion for pharmaceutical research. Among them:
Molecular Dynamics Simulation for Studying Protein-Ligand Interactions.  Molecular dynamics (MD) is a computational method employed to simulate the movement and behavior of molecules over time. Specifically in the context of protein-ligand interactions, MD offers a powerful tool for understanding the dynamic nature of these complexes at the atomic level. By solving Newton’s equations of motion numerically, MD simulations can elucidate how proteins and ligands interact, providing insights into binding affinities, structural changes, and dynamic fluctuations. Through MD, researchers can observe the complex interplay between proteins and ligands, capturing the subtle nuances of their interactions, which are often crucial for drug design and optimization. By simulating the trajectories of individual atoms, MD enables the exploration of conformational changes and energetics, offering invaluable information for rational drug discovery efforts.
Molecular dynamics (MD) is a computational method employed to simulate the movement and behavior of molecules over time. Specifically in the context of protein-ligand interactions, MD offers a powerful tool for understanding the dynamic nature of these complexes at the atomic level. By solving Newton’s equations of motion numerically, MD simulations can elucidate how proteins and ligands interact, providing insights into binding affinities, structural changes, and dynamic fluctuations. Through MD, researchers can observe the complex interplay between proteins and ligands, capturing the subtle nuances of their interactions, which are often crucial for drug design and optimization. By simulating the trajectories of individual atoms, MD enables the exploration of conformational changes and energetics, offering invaluable information for rational drug discovery efforts.
Understanding the dynamic balance between Protein Kinases’ Confirmations. 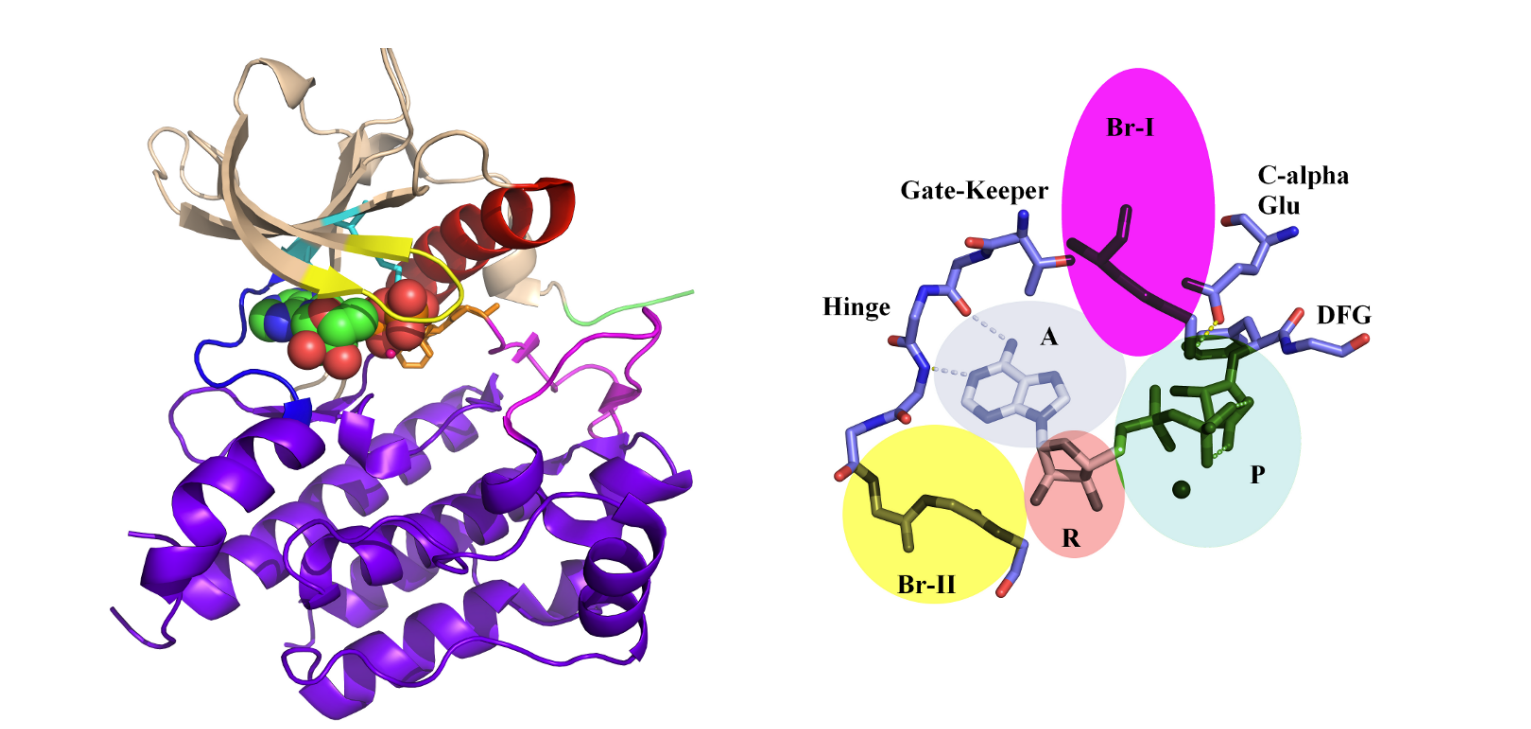 Kinases, crucial enzymes regulating cellular signaling pathways, often exist in two distinct conformations: active and inactive. The dynamic equilibrium between these states plays a pivotal role in cellular function. In the active conformation, kinases are poised to phosphorylate substrates, facilitating signal transduction. This state is typically characterized by an open conformation of the catalytic site, allowing substrate access and efficient catalysis.
Kinases, crucial enzymes regulating cellular signaling pathways, often exist in two distinct conformations: active and inactive. The dynamic equilibrium between these states plays a pivotal role in cellular function. In the active conformation, kinases are poised to phosphorylate substrates, facilitating signal transduction. This state is typically characterized by an open conformation of the catalytic site, allowing substrate access and efficient catalysis.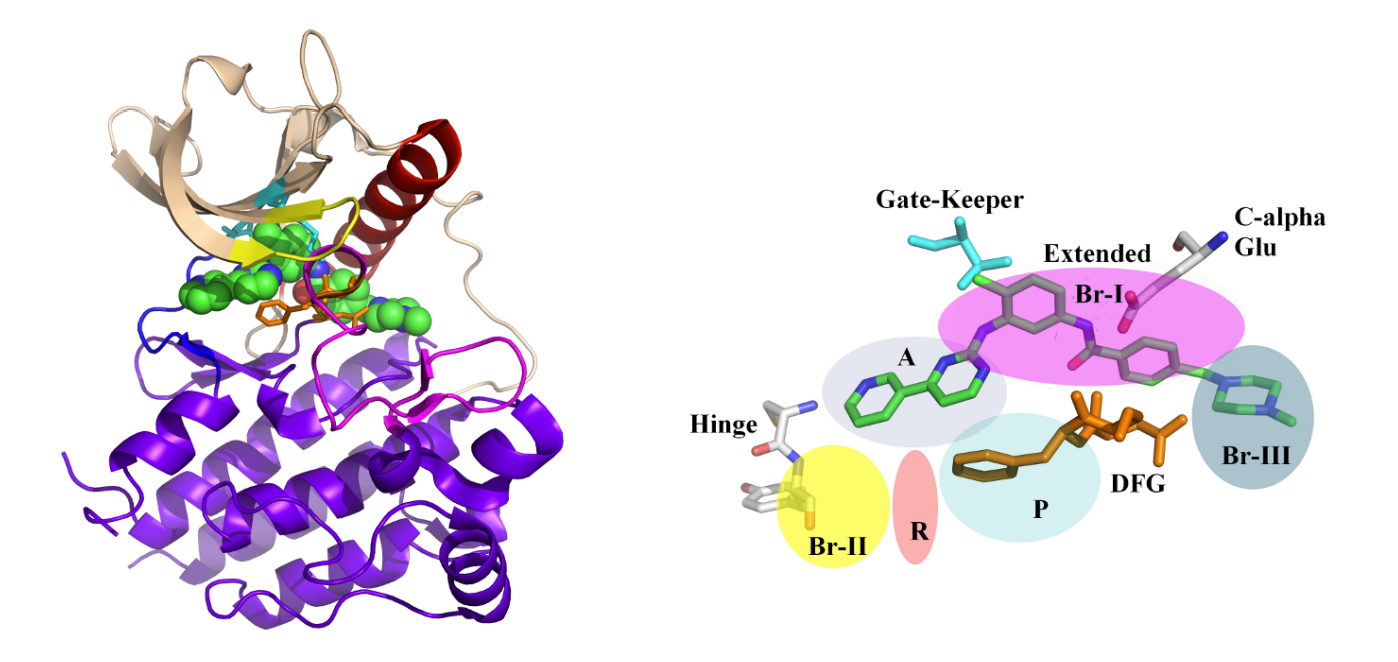 Conversely, in the inactive conformation, the catalytic site is often inaccessible or adopts a less favorable geometry for substrate binding and phosphorylation. This dynamic balance between active and inactive states is finely tuned by various factors, including allosteric regulation, post-translational modifications, and interactions with regulatory proteins. Understanding and manipulating this equilibrium is crucial for therapeutic intervention, as dysregulation of kinase activity is implicated in numerous diseases, including cancer and neurodegenerative disorders. —
Conversely, in the inactive conformation, the catalytic site is often inaccessible or adopts a less favorable geometry for substrate binding and phosphorylation. This dynamic balance between active and inactive states is finely tuned by various factors, including allosteric regulation, post-translational modifications, and interactions with regulatory proteins. Understanding and manipulating this equilibrium is crucial for therapeutic intervention, as dysregulation of kinase activity is implicated in numerous diseases, including cancer and neurodegenerative disorders. —
Computational Simulation for Spectroscopy Computational simulation plays a pivotal role in understanding complex processes such as EI (Electron Ionization) fragmentation in mass spectrometry.  By employing computational methods such as molecular dynamics or quantum mechanics-based simulations, researchers can model the energetic pathways of molecular fragmentation upon electron ionization, providing insights into the fragmentation patterns observed in mass spectra. These simulations allow for the prediction of fragment ions and their abundances, aiding in the identification and characterization of unknown compounds. Similarly, in quantum simulation of spectroscopy, computational methods such as density functional theory (DFT) or coupled cluster theory are utilized to model the electronic structure and spectroscopic properties of molecules. These simulations provide valuable information about molecular energy levels, transition probabilities, and spectra, aiding in the interpretation and prediction of experimental spectroscopic data across various spectroscopic techniques, including UV-visible, infrared, and NMR spectroscopy. Through computational simulation, researchers can gain a deeper understanding of the underlying physics governing spectroscopic phenomena, enabling more accurate interpretation and prediction of experimental results.
By employing computational methods such as molecular dynamics or quantum mechanics-based simulations, researchers can model the energetic pathways of molecular fragmentation upon electron ionization, providing insights into the fragmentation patterns observed in mass spectra. These simulations allow for the prediction of fragment ions and their abundances, aiding in the identification and characterization of unknown compounds. Similarly, in quantum simulation of spectroscopy, computational methods such as density functional theory (DFT) or coupled cluster theory are utilized to model the electronic structure and spectroscopic properties of molecules. These simulations provide valuable information about molecular energy levels, transition probabilities, and spectra, aiding in the interpretation and prediction of experimental spectroscopic data across various spectroscopic techniques, including UV-visible, infrared, and NMR spectroscopy. Through computational simulation, researchers can gain a deeper understanding of the underlying physics governing spectroscopic phenomena, enabling more accurate interpretation and prediction of experimental results.
More Related Research Topics:
- Tackling the challenge of Mastocytosis/GIST’s targeted therapy resistance driven by the mutant D816V c-KIT Tyrosine kinase.
- Unraveling the intricacies of Kinase inhibitors’ selectivity, particularly against GSK3beta/CDK2 kinases.
- Employing fast physics-based methodologies like Linear Interaction Energy (LIE) and MM-PBSA to delve into the dynamics of Protein Kinases.
- Utilizing virtual screening techniques to unearth potential enzyme inhibitors, pushing the boundaries of drug discovery.
- Spearheading the discovery and refinement of Histone-Acetyltransferase (HAT) inhibitors, driven by a relentless pursuit of innovation.
- Pioneering the identification and enhancement of novel inhibitors targeting mPGES-1, Acetyl-Choline-Esterase, and Tyrosinase.
- Leveraging structure-based methods such as Docking and Molecular Dynamics to gain insights into molecular interactions.
- Exploring the realm of drug delivery systems, controlled release mechanisms, and polymer sciences for enhanced therapeutic outcomes.
- Navigating the complexities of Cheminformatics, Chemometrics, and chemical data management to extract meaningful insights.
- Harnessing the power of R, Python, and SQL for robust data processing and analysis.
- Mastering chromatography, spectrometry, and analytical chemistry techniques to validate and refine our discoveries.